3. Flexible fairness for batch correction
3.1. Latent combination tutorial for erythroid organ inference
3.1.1. The model that will be loaded in the following cells, has been trained on the Suo et al. dataset using the following script:
[1]:
# enable autoreload
%load_ext autoreload
%autoreload 2
%matplotlib inline
import os
import random
import scvi
scvi.settings.seed = 0
import scanpy as sc
import anndata as ad
import torch
import numpy as np
import pandas as pd
import gc
torch.set_float32_matmul_precision('medium')
import matplotlib.pyplot as plt
from matplotlib.pyplot import rc_context
import seaborn as sns
import warnings
from tqdm import tqdm
warnings.simplefilter("ignore", UserWarning)
warnings.filterwarnings("ignore")
from celldisect import CellDISECT
Global seed set to 0
Global seed set to 0
[2]:
data_path = '/lustre/scratch126/cellgen/team205/aa34/Arian/Dis2P/dis2p_reproducibility/figure_notebooks/antony/suo_prep_data.h5ad'
adata = sc.read_h5ad(data_path)
adata.X = adata.layers['counts'].copy()
adata = adata[adata.X.sum(1) != 0].copy()
adata.var['highly_variable'] = adata.var['highly_variable'].astype(bool)
cats = ['integration_donor', 'integration_library_platform_coarse',
'organ']
cell_type_included = False # Set to True if you have provided a cell type annotation in the cats list
if not cell_type_included:
adata.obs["_cluster"] = (
"0" # Dummy obs for inference (not-training) time, to avoid computing neighbors and clusters again in setup_anndata | AVOID ADDING BEFORE TRAINING
)
adata
[2]:
AnnData object with n_obs × n_vars = 841966 × 8192
obs: 'study', 'sample_ID', 'lane_ID', 'organ', 'age', 'cell_type', 'sex', 'sex_inferred', 'sample_status', 'library_platform', 'strand_sequence', 'biological_unit', 'doi', 'institute', 'study_PI', 'subject_ID', 'ethnicity_1', 'ethnicity_2', 'disease_known', 'disease_condition', 'subject_type', 'sample_type', 'sample_cultured', 'anatomical_region', 'anatomical_region_level_2', 'protocol_tissue_dissociation', 'cell_enrichment', 'reference_genome', 'reference_genome_ensembl_release', 'reads_processing', 'sequencing_platform', 'Sample_orig_author', 'Anatomical_region_orig_author', 'Termination_method_orig_author', 'Method_orig_author', 'anno_lvl_2_final_clean_orig_author', 'integration_donor', 'integration_biological_unit', 'integration_sample_status', 'integration_library_platform_coarse', 'author_batch', 'Chuan_meta_Original_Label', 'Chuan_meta_Harmonised_Label', 'Chuan_meta_Tissue', 'Chuan_meta_Technology', 'Chuan_meta_Donor', 'Chuan_meta_Age', 'orig_anno', 'trimester', 'bin_age', '_cluster', '_scvi_batch', '_scvi_labels', '_scvi_cluster'
var: 'GeneID', 'GeneName', 'n_counts', 'highly_variable', 'means', 'dispersions', 'dispersions_norm'
uns: '_scvi_manager_uuid', '_scvi_uuid', 'bin_age_colors', 'cell_type_colors', 'hvg', 'integration_donor_colors', 'integration_library_platform_coarse_colors', 'log1p', 'organ_colors'
obsm: '_scvi_extra_categorical_covs'
layers: 'counts'
[3]:
pd.set_option('display.max_rows', 500)
adata.obs['orig_anno'].value_counts()
[3]:
orig_anno
MID_ERY 55104
MACROPHAGE_IRON_RECYCLING 48640
MACROPHAGE_LYVE1_HIGH 35001
NK 30284
FIBROBLAST_VII 30142
FIBROBLAST_I 25041
DP(P)_T 22839
FIBROBLAST_IV 22634
YS_ERY 20370
MATURE_B 17489
MACROPHAGE_MHCII_HIGH 14872
MACROPHAGE_PROLIFERATING 14391
DN(Q)_T 13655
CD4+T 13649
FIBROBLAST_VIII 12719
ENDOTHELIUM_IV 12601
DP(Q)_T 12390
EARLY_ERY 11897
MONOCYTE_I_CXCR4 11891
DC2 11491
FIBROBLAST_X 11415
MYOFIBROBLAST 11238
FIBROBLAST_II 10984
LATE_ERY 10814
MONOCYTE_II_CCR2 10660
LARGE_PRE_B 10468
PROMONOCYTE 9866
DN(P)_T 9810
ENDOTHELIUM_III 9416
CD8+T 9369
EPITHELIUM_II 9155
ENDOTHELIUM_I 8972
FIBROBLAST_XI 8864
MONOCYTE_III_IL1B 8675
SMALL_PRE_B 8263
PRO_B 8113
CYCLING_EPITHELIUM 7881
CYCLING_FIBROBLAST_I 7870
HEPATOCYTE_II 7845
CYCLING_FIBROBLAST_II 7770
ABT(ENTRY) 7066
CYCLING_NK 7052
TREG 6850
LATE_PRO_B 6340
MUSCLE_SATELLITE 6041
ILC3 5931
EARLY_MK 5831
MEP 5729
DEVELOPING_NEPHRON_I 5727
TYPE_1_INNATE_T 5727
B1 5652
FIBROBLAST_V 5564
MYOFIBROBLAST_I 5562
LATE_MK 5472
FIBROBLAST_XIV 5333
MACROPHAGE_KUPFFER_LIKE 5214
CD8AA 4737
FIBROBLAST_III 4569
FIBROBLAST_VI 4300
GMP 3995
CYCLING_DC 3908
MAST_CELL 3897
HSC_MPP 3816
MOP 3753
CYCLING_YS_ERY 3516
MACROPHAGE_ERY 3302
FIBROBLAST_IX 3286
DC1 3138
PROMYELOCYTE 3113
PRE_PRO_B 3034
TYPE_3_INNATE_T 3031
CYCLING_T 2985
SMOOTH_MUSCLE 2912
FIBROBLAST_XIII 2892
FIBROBLAST_XV 2804
FIBROBLAST_XII 2719
IMMATURE_B 2689
EPITHELIUM_I 2597
CMP 2579
HEPATOCYTE_I 2561
PDC 2473
CYCLING_MPP 2440
FIBROBLAST_XVII 2342
EOSINOPHIL_BASOPHIL 2334
MEMP 2238
HEPATOCYTE-LIKE 2224
KERATINOCYTE 2151
NEURON 2010
CYCLING_B 1909
DEVELOPING_NEPHRON_II 1749
OSTEOBLAST 1734
DN(early)_T 1670
LMPP_MLP 1395
MACROPHAGE_PERI 1301
ENDOTHELIUM_II 1244
OSTEOCLAST 1199
NEUTROPHIL 1075
MESOTHELIUM 1039
MYELOCYTE 1019
CYCLING_MEMP 922
MELANOCYTE 899
MACROPHAGE_TREM2 888
ENDOTHELIUM_V 809
CYCLING_ILC 541
CYCLING_PDC 532
ENTEROENDOCRINE_I 503
ILC2 468
MIGRATORY_DC 421
INTERSTITIAL_CELLS_OF_CAJAL 409
YS_STROMA 334
MESENCHYMAL_LYMPHOID_TISSUE_ORGANISER 300
AS_DC 266
CHONDROCYTE 253
ENTEROENDOCRINE_II 231
PRE_DC2 215
FIBROBLAST_XVI 207
DC_PROGENITOR 200
SKELETAL_MUSCLE 123
LANGERHANS_CELLS 66
PLASMA_B 61
Name: count, dtype: int64
[4]:
celltypes = [
'MID_ERY',
'EARLY_ERY',
'LATE_ERY',
'HSC_MPP',
'MEMP',
'CYCLING_MEMP',
'MEP',
]
[5]:
adata = adata[adata.obs['orig_anno'].isin(celltypes)].copy()
[6]:
module_name = f'Antony_suoFull_NoAge'
pre_path = f'/lustre/scratch126/cellgen/team205/aa34/Arian/Dis2P/models/{module_name}'
model_name = 'development_clustering_pretrainAE_0_maxEpochs_1000_reconW_20_cfWeight_0.8_beta_0.003_clf_0.02_adv_0.014_advp_5_n_cf_1_lr_0.003_wd_5e-05_new_cf_True_dropout_0.1_n_hidden_128_n_latent_32_n_layers_2_weighted_classifier_False'
model = CellDISECT.load(f"{pre_path}/{model_name}", adata=adata)
INFO File
/lustre/scratch126/cellgen/team205/aa34/Arian/Dis2P/models/Antony_suoFull_NoAge/development_clustering_pre
trainAE_0_maxEpochs_1000_reconW_20_cfWeight_0.8_beta_0.003_clf_0.02_adv_0.014_advp_5_n_cf_1_lr_0.003_wd_5e
-05_new_cf_True_dropout_0.1_n_hidden_128_n_latent_32_n_layers_2_weighted_classifier_False/model.pt already
downloaded
Cluster covariate already present in adata.obs, remove in case you want to re-run, skipping!
[7]:
adata
[7]:
AnnData object with n_obs × n_vars = 90520 × 8192
obs: 'study', 'sample_ID', 'lane_ID', 'organ', 'age', 'cell_type', 'sex', 'sex_inferred', 'sample_status', 'library_platform', 'strand_sequence', 'biological_unit', 'doi', 'institute', 'study_PI', 'subject_ID', 'ethnicity_1', 'ethnicity_2', 'disease_known', 'disease_condition', 'subject_type', 'sample_type', 'sample_cultured', 'anatomical_region', 'anatomical_region_level_2', 'protocol_tissue_dissociation', 'cell_enrichment', 'reference_genome', 'reference_genome_ensembl_release', 'reads_processing', 'sequencing_platform', 'Sample_orig_author', 'Anatomical_region_orig_author', 'Termination_method_orig_author', 'Method_orig_author', 'anno_lvl_2_final_clean_orig_author', 'integration_donor', 'integration_biological_unit', 'integration_sample_status', 'integration_library_platform_coarse', 'author_batch', 'Chuan_meta_Original_Label', 'Chuan_meta_Harmonised_Label', 'Chuan_meta_Tissue', 'Chuan_meta_Technology', 'Chuan_meta_Donor', 'Chuan_meta_Age', 'orig_anno', 'trimester', 'bin_age', '_cluster', '_scvi_batch', '_scvi_labels', '_scvi_cluster'
var: 'GeneID', 'GeneName', 'n_counts', 'highly_variable', 'means', 'dispersions', 'dispersions_norm'
uns: '_scvi_manager_uuid', '_scvi_uuid', 'bin_age_colors', 'cell_type_colors', 'hvg', 'integration_donor_colors', 'integration_library_platform_coarse_colors', 'log1p', 'organ_colors'
obsm: '_scvi_extra_categorical_covs'
layers: 'counts'
3.3. You can also use the combination of covariates/latents as you like, here we are using a combination/concatenation of Z0 with each of the covariates to make new latents Z0+ZCov
[9]:
# Z_{0+Z_i} = Z_0 + Z_i | Optional
# This represents the combination of latents with Z_0.
# Here, Z_i is a vector where all elements are zero except for the i-th section.
# Z_0 is [latent_0, 0, ..., 0] and Z_i is [0, ..., 0, latent_i, 0, ..., 0].
# When mixing two latents, it will look like [latent_0, 0, latent_i, 0, ..., 0].
for i in range(len(cats)): # loop over all Z_i
label = cats[i]
latent_name = f"CellDISECT_Z_0+{label}"
adata.obsm[latent_name] = (
adata.obsm["CellDISECT_Z_0"].copy() + adata.obsm[f"CellDISECT_Z_{label}"].copy()
)
3.3.1. With the same pattern as above, you can mix any combination of covariates and latents of your choosing:
[10]:
latent_to_make = ['0', 'organ', 'integration_donor']
# or
# latent_to_make = ['organ', 'integration_donor']
# or any other combination
latent_name = "CellDISECT_Z_" + "_".join(latent_to_make)
temp_latent = np.zeros_like(adata.obsm["CellDISECT_Z_0"])
for name in latent_to_make:
temp_latent += adata.obsm[f"CellDISECT_Z_{name}"].copy()
adata.obsm[latent_name] = (
temp_latent.copy()
)
del temp_latent
[11]:
latent_name
[11]:
'CellDISECT_Z_0_organ_integration_donor'
You can see that the latent for CellDISECT_Z_0_organ_integration_donor, has the latent vectors in their respective indices, and zeros in the integration_library_platform_coarse indices which was not selected
[12]:
adata.obsm[latent_name][:, :128].sum(0)
[12]:
array([-1.75040391e+04, -3.38555938e+04, -1.87798398e+04, 9.55953320e+03,
4.61457773e+04, 9.14440703e+04, 7.54916064e+03, -2.18722375e+05,
-4.83425273e+04, -1.10249328e+05, 3.78672344e+04, -1.38188877e+04,
3.27485781e+04, 3.94175098e+03, 3.88769375e+04, 5.67719922e+04,
1.08032750e+05, -2.51776611e+02, 1.60374854e+04, -6.89519688e+04,
3.04145332e+04, -5.89433750e+04, -5.21918789e+04, -3.42050547e+04,
-1.01287549e+04, 5.39129736e+03, -1.24341914e+05, 1.29704133e+05,
-1.53636025e+04, 4.29150156e+04, 5.20313281e+04, 1.09389268e+04,
-4.40014404e+02, 8.01885437e+02, 9.21002930e+02, -5.22604102e+03,
-1.21806416e+04, 1.41498975e+03, -5.41523389e+03, 2.66045868e+02,
-1.32705566e+03, -1.68192981e+03, -2.51068730e+04, -1.28214954e+03,
-1.68837540e+02, 4.64866914e+04, -2.78392749e+03, 7.40726547e+01,
9.00216614e+02, 3.55316382e+03, -1.16640845e+03, 2.92287988e+03,
-4.66438330e+03, 2.16277206e+02, 3.64461680e+04, 8.10476406e+04,
5.75590703e+04, -2.93583081e+03, -7.71643982e+02, 1.16263086e+04,
9.97728882e+02, 1.84864197e+03, -6.83654297e+04, 1.75080429e+02,
0.00000000e+00, 0.00000000e+00, 0.00000000e+00, 0.00000000e+00,
0.00000000e+00, 0.00000000e+00, 0.00000000e+00, 0.00000000e+00,
0.00000000e+00, 0.00000000e+00, 0.00000000e+00, 0.00000000e+00,
0.00000000e+00, 0.00000000e+00, 0.00000000e+00, 0.00000000e+00,
0.00000000e+00, 0.00000000e+00, 0.00000000e+00, 0.00000000e+00,
0.00000000e+00, 0.00000000e+00, 0.00000000e+00, 0.00000000e+00,
0.00000000e+00, 0.00000000e+00, 0.00000000e+00, 0.00000000e+00,
0.00000000e+00, 0.00000000e+00, 0.00000000e+00, 0.00000000e+00,
7.52727832e+03, 5.47131836e+03, 1.43137451e+04, 3.71644946e+03,
3.35054844e+04, -4.50400156e+04, 1.40292125e+05, -7.29384062e+04,
-9.61884766e+03, -3.87217432e+03, -5.54280090e+02, -4.60546289e+03,
2.69948164e+04, -5.79350049e+03, 6.75064648e+03, -9.11916309e+03,
3.27080322e+03, 6.99119688e+04, 8.06322021e+03, 8.51738770e+03,
3.85352148e+04, 4.99245020e+03, 1.32849082e+04, 6.24061914e+04,
-3.24508887e+04, -7.79491016e+03, 7.29276719e+04, -5.01927124e+02,
-2.96022344e+04, -7.79007568e+03, 6.99948145e+03, 5.96621367e+04],
dtype=float32)
[13]:
cats
[13]:
['integration_donor', 'integration_library_platform_coarse', 'organ']
We are computing the neighbors and umaps for each of these latents, the first loop computes these for Z_0 and Z_organ, and the second look does it for Z_0+organ. (You can loop over all covariates, we hardcoded these 2 latents here ([0, 3]) for faster computation.
[14]:
# Compute neighbors and UMAPs for the latent representations (this might take a while, consider running it using RAPIDS scanpy with a GPU if data is large)
for i in [0, 3]: # loop over Z_0 and Z_organ | Neighbors and UMAPs for Z_0 and Z_organ
if i == 0:
latent_name = f"CellDISECT_Z_{i}"
else:
label = cats[i - 1]
latent_name = f"CellDISECT_Z_{label}"
latent = ad.AnnData(X=adata.obsm[f"{latent_name}"], obs=adata.obs)
sc.pp.neighbors(adata=latent, use_rep="X")
sc.tl.umap(adata=latent)
adata.uns[f"{latent_name}_neighbors"] = latent.uns["neighbors"]
adata.obsm[f"{latent_name}_umap"] = latent.obsm["X_umap"]
gc.collect()
for i in [2]: # loop over Z_organ | Neighbors and UMAPs for Z_0+Z_organ | Optional
label = cats[i]
latent_name = f"CellDISECT_Z_0+{label}"
latent = ad.AnnData(X=adata.obsm[f"{latent_name}"], obs=adata.obs)
sc.pp.neighbors(adata=latent, use_rep="X")
sc.tl.umap(adata=latent)
adata.uns[f"{latent_name}_neighbors"] = latent.uns["neighbors"]
adata.obsm[f"{latent_name}_umap"] = latent.obsm["X_umap"]
gc.collect()
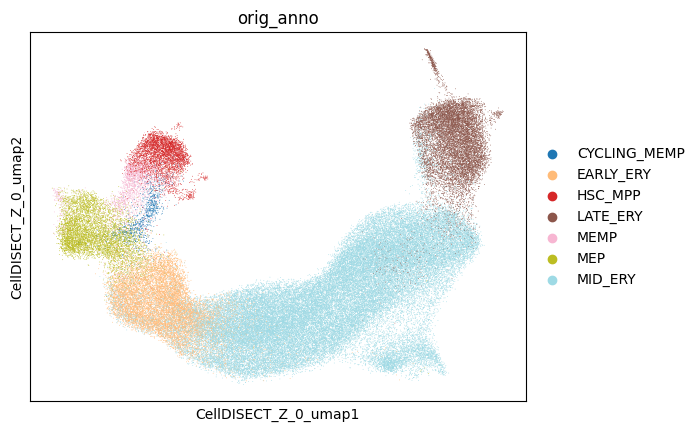
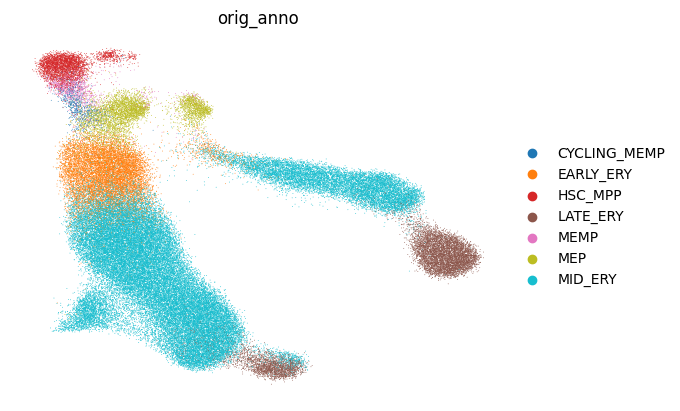
You can see the separation of cell types in Z0 since it was not provided as a covariate and CellDISECT has learnt it unsupervised
[15]:
fig, ax = plt.subplots(nrows=1, ncols=1)
sc.pl.embedding(
adata,
basis='CellDISECT_Z_0_umap',
color='orig_anno',
groups=celltypes,
palette='tab20',
ax=ax,
show=False,
)
# fig.savefig('ery_figs/z0_orig_anno.png', dpi=150, bbox_inches='tight')
# fig.savefig('ery_figs/z0_orig_anno.svg', dpi=150, bbox_inches='tight')
# plt.show()
# plt.close(fig)
[15]:
<Axes: title={'center': 'orig_anno'}, xlabel='CellDISECT_Z_0_umap1', ylabel='CellDISECT_Z_0_umap2'>
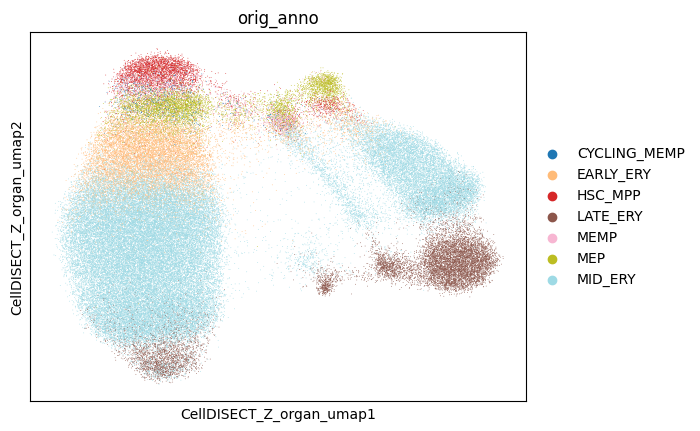
[16]:
fig, ax = plt.subplots(nrows=1, ncols=1)
sc.pl.embedding(
adata,
basis='CellDISECT_Z_organ_umap',
color='orig_anno',
groups=celltypes,
ax=ax,
show=False,
)
# fig.savefig('ery_figs/zOrgan_orig_anno.png', dpi=150, bbox_inches='tight')
# fig.savefig('ery_figs/zOrgan_orig_anno.svg', dpi=150, bbox_inches='tight')
# plt.show()
# plt.close(fig)
[16]:
<Axes: title={'center': 'orig_anno'}, xlabel='CellDISECT_Z_organ_umap1', ylabel='CellDISECT_Z_organ_umap2'>
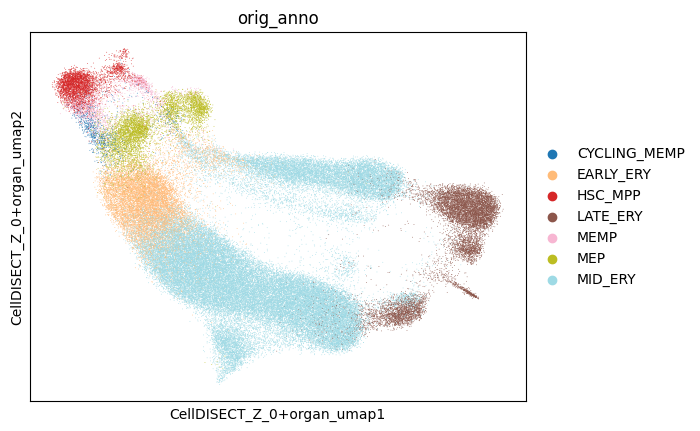
3.3.2. You can easily see the separation of different organs and cell types within one latent represantation (Z0+Organ) and you can analyse the different clusters
[17]:
fig, ax = plt.subplots(nrows=1, ncols=1)
sc.pl.embedding(
adata, basis='CellDISECT_Z_0+organ_umap', color='orig_anno', groups=celltypes,
ax=ax,
show=False,
)
# fig.savefig('ery_figs/zOrgan+0_orig_anno.png', dpi=150, bbox_inches='tight')
# fig.savefig('ery_figs/zOrgan+0_orig_anno.svg', dpi=150, bbox_inches='tight')
# plt.show()
# plt.close(fig)
[17]:
<Axes: title={'center': 'orig_anno'}, xlabel='CellDISECT_Z_0+organ_umap1', ylabel='CellDISECT_Z_0+organ_umap2'>
[18]:
fig, ax = plt.subplots(nrows=1, ncols=1)
sc.pl.embedding(
adata, basis='CellDISECT_Z_0+organ_umap', color='organ',
ax=ax,
show=False,
)
# fig.savefig('ery_figs/zOrgan+0_organ.png', dpi=150, bbox_inches='tight')
# fig.savefig('ery_figs/zOrgan+0_organ.svg', dpi=150, bbox_inches='tight')
# plt.show()
# plt.close(fig)
[18]:
<Axes: title={'center': 'organ'}, xlabel='CellDISECT_Z_0+organ_umap1', ylabel='CellDISECT_Z_0+organ_umap2'>
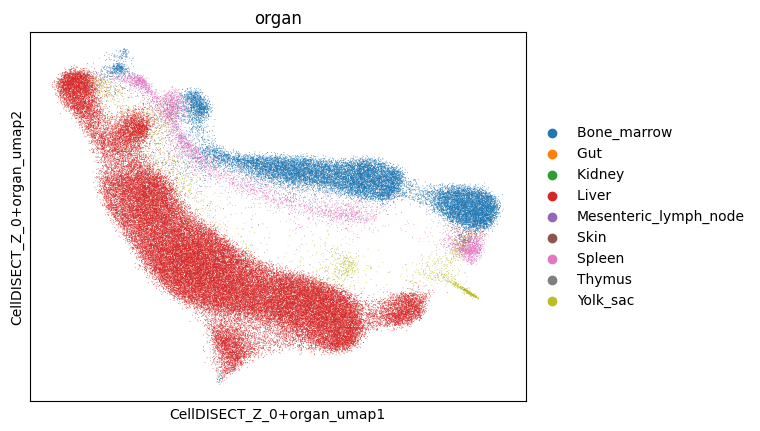
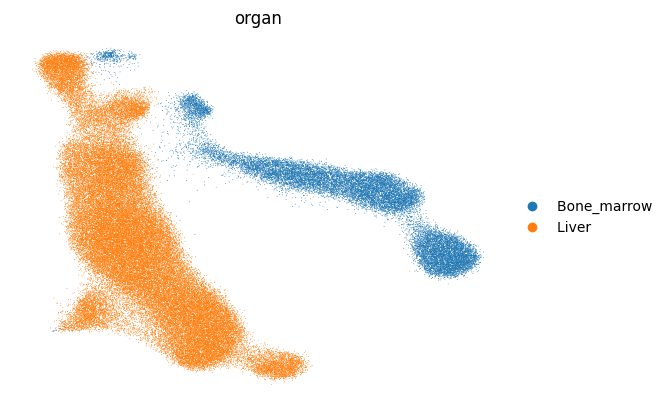
3.4. Now we want to take a closer look at Liver and Bone Marrow cells
[19]:
adata = adata[adata.obs['organ'].isin(['Liver ', 'Bone_marrow '])].copy()
adata
[19]:
AnnData object with n_obs × n_vars = 82585 × 8192
obs: 'study', 'sample_ID', 'lane_ID', 'organ', 'age', 'cell_type', 'sex', 'sex_inferred', 'sample_status', 'library_platform', 'strand_sequence', 'biological_unit', 'doi', 'institute', 'study_PI', 'subject_ID', 'ethnicity_1', 'ethnicity_2', 'disease_known', 'disease_condition', 'subject_type', 'sample_type', 'sample_cultured', 'anatomical_region', 'anatomical_region_level_2', 'protocol_tissue_dissociation', 'cell_enrichment', 'reference_genome', 'reference_genome_ensembl_release', 'reads_processing', 'sequencing_platform', 'Sample_orig_author', 'Anatomical_region_orig_author', 'Termination_method_orig_author', 'Method_orig_author', 'anno_lvl_2_final_clean_orig_author', 'integration_donor', 'integration_biological_unit', 'integration_sample_status', 'integration_library_platform_coarse', 'author_batch', 'Chuan_meta_Original_Label', 'Chuan_meta_Harmonised_Label', 'Chuan_meta_Tissue', 'Chuan_meta_Technology', 'Chuan_meta_Donor', 'Chuan_meta_Age', 'orig_anno', 'trimester', 'bin_age', '_cluster', '_scvi_batch', '_scvi_labels', '_scvi_cluster'
var: 'GeneID', 'GeneName', 'n_counts', 'highly_variable', 'means', 'dispersions', 'dispersions_norm'
uns: '_scvi_manager_uuid', '_scvi_uuid', 'bin_age_colors', 'cell_type_colors', 'hvg', 'integration_donor_colors', 'integration_library_platform_coarse_colors', 'log1p', 'organ_colors', 'CellDISECT_Z_0_neighbors', 'CellDISECT_Z_organ_neighbors', 'CellDISECT_Z_0+organ_neighbors', 'orig_anno_colors'
obsm: '_scvi_extra_categorical_covs', 'CellDISECT_Z_0', 'CellDISECT_Z_integration_donor', 'CellDISECT_Z_not_integration_donor', 'CellDISECT_Z_integration_library_platform_coarse', 'CellDISECT_Z_not_integration_library_platform_coarse', 'CellDISECT_Z_organ', 'CellDISECT_Z_not_organ', 'CellDISECT_Z_0+integration_donor', 'CellDISECT_Z_0+integration_library_platform_coarse', 'CellDISECT_Z_0+organ', 'CellDISECT_Z_0_organ_integration_donor', 'CellDISECT_Z_0_umap', 'CellDISECT_Z_organ_umap', 'CellDISECT_Z_0+organ_umap'
layers: 'counts'
3.4.1. We recompute the neighbors for this subset
[20]:
anno_col = 'orig_anno'
latent = 'CellDISECT_Z_0+organ'
sc.pp.neighbors(adata, n_neighbors=15, use_rep=latent)
sc.tl.umap(adata)
[21]:
fig, ax = plt.subplots(nrows=1, ncols=1)
sc.pl.umap(
adata,
color=anno_col,
palette='tab10',
frameon=False,
ax=ax,
show=False,
)
# fig.savefig('ery_figs/subset_zrgan+0_organ.png', dpi=150, bbox_inches='tight')
# fig.savefig('ery_figs/subset_zrgan+0_organ.svg', dpi=150, bbox_inches='tight')
# plt.show()
# plt.close(fig)
[21]:
<Axes: title={'center': 'orig_anno'}, xlabel='UMAP1', ylabel='UMAP2'>
[37]:
fig, ax = plt.subplots(nrows=1, ncols=1)
sc.pl.umap(
adata,
color='organ',
frameon=False,
ax=ax,
show=True,
)
# fig.savefig('ery_figs/subset_zrgan+0_organ.png', dpi=150, bbox_inches='tight')
# fig.savefig('ery_figs/subset_zrgan+0_organ.svg', dpi=150, bbox_inches='tight')
# plt.show()
# plt.close(fig)
[22]:
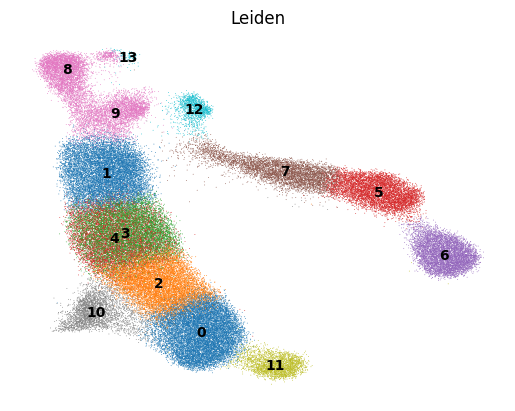
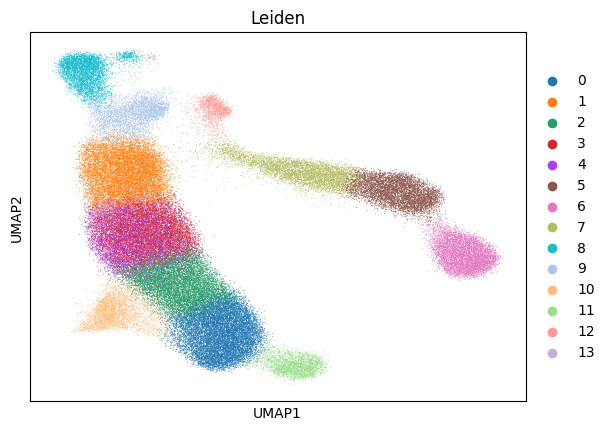
sc.tl.leiden(adata, key_added='Leiden', resolution = 2)
sc.pl.umap(adata, color='Leiden')
[23]:
fig, ax = plt.subplots(nrows=1, ncols=1)
sc.pl.umap(adata, color='Leiden', legend_loc = 'on data',
palette='tab10',
frameon=False,
ax=ax,
show=False,
)
# fig.savefig('ery_figs/subset_zrgan+0_leiden.png', dpi=150, bbox_inches='tight')
# fig.savefig('ery_figs/subset_zrgan+0_leiden.svg', dpi=150, bbox_inches='tight')
# plt.show()
# plt.close(fig)
[23]:
<Axes: title={'center': 'Leiden'}, xlabel='UMAP1', ylabel='UMAP2'>